Posted onInDeePMD-kitWord count in article: 1.7kReading time ≈6 mins.
Recently, Dr. Mei Jia from Shangqiu Normal University, Dr. Yongbin Zhuang from École Polytechnique Fédérale de Lausanne (EPFL), and Prof. Jun Cheng from Xiamen University conducted an in-depth study on the proton transfer mechanism at the SnO₂(110)/H₂O interface by combining ab initio molecular dynamics (AIMD) with the Deep Potential (DP) method. The team used AIMD to obtain the electronic structure of the interface system and applied the Deep Potential Molecular Dynamics (DPMD) model to accelerate molecular dynamics simulations, enabling larger-scale and longer-timescale simulations. This combination of methods allowed the researchers to analyze the free energy distributions of different proton transfer pathways in detail and to reveal the influence of the solvation environment on the proton transfer process.
The related findings have been published in the high-impact journal Precision Chemistry, under the title “Water-Mediated Proton Hopping Mechanisms at the SnO₂(110)/H₂O Interface from Ab Initio Deep Potential Molecular Dynamics.” Dr. Mei Jia and Dr. Yongbin Zhuang are the co-first authors, and Prof. Jun Cheng is the corresponding author.
Posted onInTutorials@NotebooksWord count in article: 737Reading time ≈3 mins.
This Notebook will approach DeePKS from an application perspective, using the perovskite system as a case study. It systematically presents the complete process of DeePKS model training and deployment, including:
Preparation of labeled data for the example system,
Model training, and
Result analysis.
Check out here: https://bohrium.dp.tech/collections/6242632852/
Tutorial Structure
Following a progression from simple to complex, this tutorial series is designed to guide readers step by step in learning DeePKS:
Single-element systems:
Start with energy label training for systems containing the same type of element.
Multi-label training for single-element systems:
Expand to training multiple labels (e.g., energy, forces, stress, and band structure) for single-element systems.
Real-world research systems:
Transition to complex research systems (e.g., those with diverse elemental compositions), incorporating multi-label training for energy, forces, stress, and band structure.
Learning Outcomes
Through this tutorial, readers will:
Gain a deep understanding of the DeePKS method,
Master how to apply it to actual model training and deployment, and
Equip themselves with essential skills to support future research.
Background
First-Principles Calculations Based on KS-DFT
First-principles calculations based on Kohn−Sham Density Functional Theory (KS-DFT) have become one of the most widely used quantum mechanical methods at the atomic and molecular scales in recent decades.
The accuracy of KS-DFT is determined by the precision of the unknown terms in the total energy—namely, the exchange-correlation functional. Among the various approximations of exchange-correlation functionals—such as LDA, GGA, meta-GGA, and hybrid functionals [1-2]—achieving a balance between accuracy and efficiency has always been a challenge.
The most commonly used functional, such as the PBE functional under the GGA approximation, performs well in terms of computational efficiency but often lacks accuracy for specific systems.
On the other hand, hybrid functionals like HSE06 offer higher accuracy but suffer from lower computational efficiency, making them impractical for handling large systems.
Opportunities with Artificial Intelligence
The rapid development of artificial intelligence (AI) has introduced new possibilities for representing and approximating high-dimensional complex functions. By leveraging deep learning models to bridge the gap between low- and high-accuracy functionals, it is now possible to achieve a good balance between efficiency and accuracy.
DeePKS Method
The DeePKS method is a deep learning-based functional correction approach developed to address this challenge [3-5]. Its key features are as follows:
Objective:
DeePKS does not reconstruct the exchange-correlation functional itself.
Instead, it uses machine learning techniques to optimize low-accuracy functionals.
How it Works:
DeePKS learns the differences in energy, forces, stress, and band structure labels between:
A baseline functional (e.g., PBE)
A target functional (e.g., HSE06)
This effectively combines the advantages of low- and high-accuracy calculations.
Key Benefits:
Good balance between efficiency and accuracy.
Low computational cost:
Correction terms are computationally as inexpensive as low-accuracy functionals.
Far less expensive than high-accuracy functionals like HSE06.
Advantages in Practical Applications
The computational cost of correction terms in DeePKS is comparable to that of low-accuracy functionals.
This makes DeePKS significantly faster than high-accuracy functionals, giving it a notable edge in practical applications.
Integration with DFT Software
During DeePKS model training, the update of model parameters alternates with the self-consistent calculations of first-principles methods. This requires DeePKS to seamlessly integrate with existing density functional theory software.
Reference: 1.Kohn W, Sham L J. Self-consistent equations including exchange and correlation effects[J]. Physical review, 1965, 140(4A): A1133. 2.Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Physical review letters, 1996, 77(18): 3865. 3.https://github.com/deepmodeling/deepks-kit/tree/develop 4.Chen Y, Zhang L, Wang H, et al. DeePKS: A comprehensive data-driven approach toward chemically accurate density functional theory[J]. Journal of Chemical Theory and Computation, 2020, 17(1): 170-181. 5.Ou Q, Tuo P, Li W, et al. DeePKS Model for Halide Perovskites with the Accuracy of a Hybrid Functional[J]. The Journal of Physical Chemistry C, 2023, 127(37): 18755-18764. 6.https://github.com/deepmodeling/abacus-develop 7.Li W, Ou Q, Chen Y, et al. DeePKS+ ABACUS as a Bridge between Expensive Quantum Mechanical Models and Machine Learning Potentials[J]. The Journal of Physical Chemistry A, 2022, 126(49): 9154-9164.
Posted onInUni-MolWord count in article: 1.5kReading time ≈6 mins.
On July 15, 2024, Bilal Aslan from the University of Cape Town, Flavio Correa da Silva from the University of São Paulo, and Geoff Nitschke from the University of Cape Town collaborated to present their research titled “Multi-Objective Evolution for Chemical Product Design” at the Genetic and Evolutionary Computation Conference (GECCO). This study introduced a chemical product design method based on multi-objective evolutionary optimization. By innovatively integrating deep learning with evolutionary algorithms, the approach optimizes molecular properties and utilizes the Uni-Mol model to evaluate molecular toxicity, providing a novel solution for the design and optimization of chemical products.
Posted onInDeepFlameWord count in article: 1.5kReading time ≈5 mins.
DeepFlame is an open-source combustion fluid dynamics platform developed for the AI for Science era [1-3], aimed at overcoming the longstanding challenges of applying traditional Computational Fluid Dynamics (CFD) in the field of combustion. Since its release, DeepFlame has garnered significant interest and attention from both academia and industry, attracting a group of outstanding developers and users. This ongoing support has provided continuous momentum for DeepFlame's development and has been a crucial driving force in its application to real-world scenarios.
In recent years, research on aerosol or spray detonation propulsion using liquid fuels has been experiencing a resurgence, and supersonic combustion, such as detonation combustion in gas-liquid two-phase systems, has been gaining increasing attention. The DeepFlame team has captured these trending topics and, based on the OpenFOAM open-source library, coupled the Euler-Lagrange model into the high-speed flow solver dfHighSpeedFoam and the low-speed flow solver dfLowMachFoam. This enables the solvers to simulate two-phase reactive flows, thereby expanding the application scenarios of DeepFlame.
Posted onInUni-MolWord count in article: 2.2kReading time ≈8 mins.
On August 19, 2024, Shuqi Lu and Zhifeng Gao from DP Technology, in collaboration with Professor Di He from Peking University, published a research article titled "Data-driven quantum chemical property prediction leveraging 3D conformations with Uni-Mol+" in Nature Communications. This study introduces Uni-Mol+, a deep learning algorithm that innovatively utilizes neural networks to iteratively optimize initial 3D molecular conformations, enabling precise prediction of quantum chemical properties. By progressively approximating Density Functional Theory (DFT) equilibrium conformations, Uni-Mol+ significantly enhances prediction accuracy, providing a powerful tool for high-throughput screening and new material design.
Posted onInDeePTBWord count in article: 1.8kReading time ≈7 mins.
In 2023, the AI for Science Institute, Beijing team introduced the v1 version of the DeePTB method, which was published on arXiv and joined the DeepModeling community. After nearly a year of rigorous peer review, it was officially published on August 8, 2024, in the international academic journal Nature Communications with the title "Deep learning tight-binding approach for large-scale electronic simulations at finite temperatures with ab initio accuracy" [1], DOI: 10.1038/s41467-024-51006-4.
The v1 version of DeePTB focuses on developing a deep learning-based method for constructing tight-binding (TB) model Hamiltonians. Based on the Slater-Koster TB parameterization, it builds first-principles equivalent electronic models using a minimal-basis set. By incorporating the localized chemical environment of atoms/bonds into the TB parameters, DeePTB achieves TB Hamiltonian predictions with near-DFT accuracy across a range of key material systems. By integrating with software like DeePMD-kit and TBPLaS, it enables the calculation and simulation of electronic structure properties and photoelectric responses in large-scale systems of up to millions of atoms in finite-temperature ensembles. This groundbreaking advancement has garnered widespread attention in the academic community and was ultimately published in Nature Communications. For more technical details on the DeePTB version, interested readers can refer to the DeePTB article in Nat Commun 15, 6772 (2024).
Posted onInUni-MolWord count in article: 2kReading time ≈7 mins.
On February 13, 2024, DP Technology published a cover article in JACS Au titled "Node-Aligned Graph-to-Graph: Elevating Template-free Deep Learning Approaches in Single-Step Retrosynthesis." This study developed a Transformer-based Node-Aligned Graph-to-Graph (NAG2G) model, significantly improving the accuracy of single-step retrosynthesis prediction.
The NAG2G model integrates 2D molecular graph and 3D conformation information, achieving atom mapping between products and reactants through node alignment. This approach overcomes the limitations of traditional template-based methods.
This groundbreaking achievement provides a powerful tool for chemical synthesis design, advancing the field of retrosynthesis and setting a new standard for single-step prediction methodologies.
Posted onInOpenLAMWord count in article: 1.1kReading time ≈4 mins.
On the journey toward developing a Large Atomic Model (LAM), the core Deep Potential development team has launched the OpenLAM initiative for the community. OpenLAM’s slogan is "Conquer the Periodic Table!" The project aims to create an open-source ecosystem centered on microscale large models, providing new infrastructure for microscopic scientific research and driving transformative advancements in microscale industrial design across fields such as materials, energy, and biopharmaceuticals.
Posted onInUni-MolWord count in article: 859Reading time ≈3 mins.
On June 17, 2024, researchers Xi Cheng and Liuqing Wen from the Shanghai Institute of Materia Medica, Chinese Academy of Sciences, in collaboration with Dingyan Wang from Lingang Laboratory, published a study titled "Highly accurate carbohydrate-binding site prediction with DeepGlycanSite" in Nature Communications [1]. This research introduces DeepGlycanSite, a deep learning-based algorithm for predicting carbohydrate-binding sites on protein structures with high precision. By leveraging Uni-Mol, DeepGlycanSite achieves exceptional accuracy in identifying carbohydrate-binding sites, providing a powerful tool for studying carbohydrate-protein interactions.
1. Research Background
Carbohydrates are widely present on the surface of all living cells, interacting with various protein families, including lectins, antibodies, enzymes, and transport proteins. These interactions regulate diverse biological processes, such as immune responses, cell differentiation, and neural development. Understanding carbohydrate-protein interactions is therefore fundamental to developing carbohydrate-based therapeutics.
However, due to the structural diversity of carbohydrates, obtaining experimental data on carbohydrate-protein interactions remains challenging. Structural determination techniques commonly used in glycobiology, such as nuclear magnetic resonance (NMR) and X-ray crystallography, require pure, stable molecules of detectable sizes.
Small carbohydrates (e.g., glucose with a molecular weight under 200 Da) are difficult to detect in structural studies due to their low atom count. On the other hand, complex long-chain carbohydrates (e.g., oligosaccharides with molecular weights exceeding 1000 Da) often involve multiple conformational states, leading to heterogeneity. In both cases, carbohydrate-binding residues of proteins cannot be clearly defined from a structural perspective.
Thus, developing a reliable tool for predicting carbohydrate-binding sites is critical to advancing our understanding of carbohydrate-protein interactions.
2. Cutting-Edge Deep Learning Technology—DeepGlycanSite
DeepGlycanSite is an equivariant graph neural network (EGNN) model based on deep learning, combining geometric features of proteins with evolutionary information to outperform state-of-the-art methods. This model not only predicts binding sites for monosaccharides and disaccharides but also accurately identifies binding sites for oligosaccharides and nucleotides.
The success of this study lies in the precise understanding of carbohydrate chemical structures, a capability significantly enhanced by Uni-Mol, which plays a critical role in the model's performance.
3. How Does Uni-Mol Assist DeepGlycanSite?
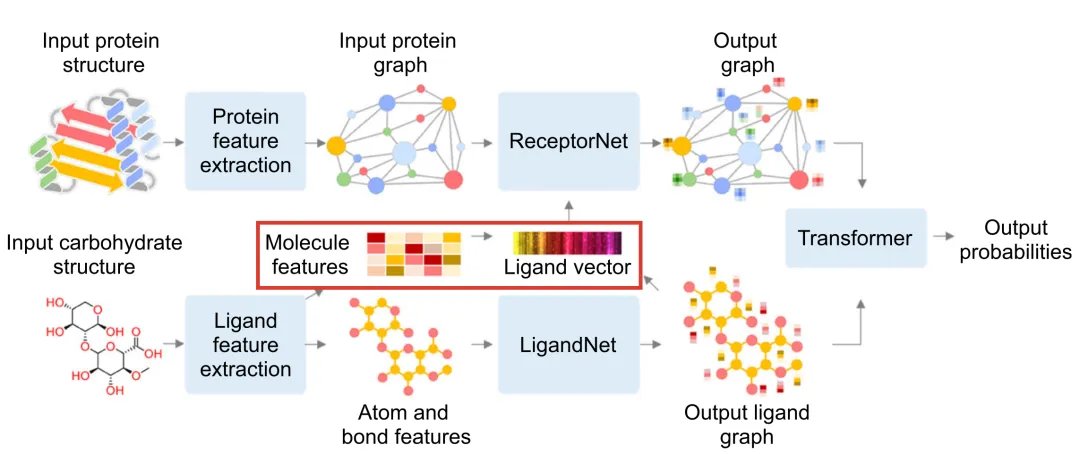
The performance of deep learning models heavily depends on the quality of feature extraction. In DeepGlycanSite, Uni-Mol is utilized to generate detailed chemical features of carbohydrates, enabling more accurate prediction of binding sites. The implementation is as follows:
3.1 Carbohydrate Processing
SMILES Representation: Rdkit is used to process the query carbohydrate and extract its SMILES representation.
Feature Generation: Uni-Mol, integrated with Rdkit, converts the SMILES representation into molecular features.
3.2 Feature Extraction
Node Features:
Include detailed atomic properties:
Atom symbol
Degree
Hybridization type
Formal charge
Number of radical electrons
Aromaticity
Total hydrogen count
Chirality
Edge Features:
Capture bond-level information:
Bond type
Conjugation
Ring membership
Stereochemical configuration
Global Molecular Features:
Generate a 512-dimensional molecular feature vector encapsulating the overall chemical information of the carbohydrate.
3.3 Feature Integration
In the DeepGlycanSite+Ligand module:
The ligand vector generated by Uni-Mol is fused with the protein graph’s node features.
This integration is processed through an attention layer for feature alignment and updating.
The combined features are then used to predict the binding probability of carbohydrates.
4. Experimental Validation and Results
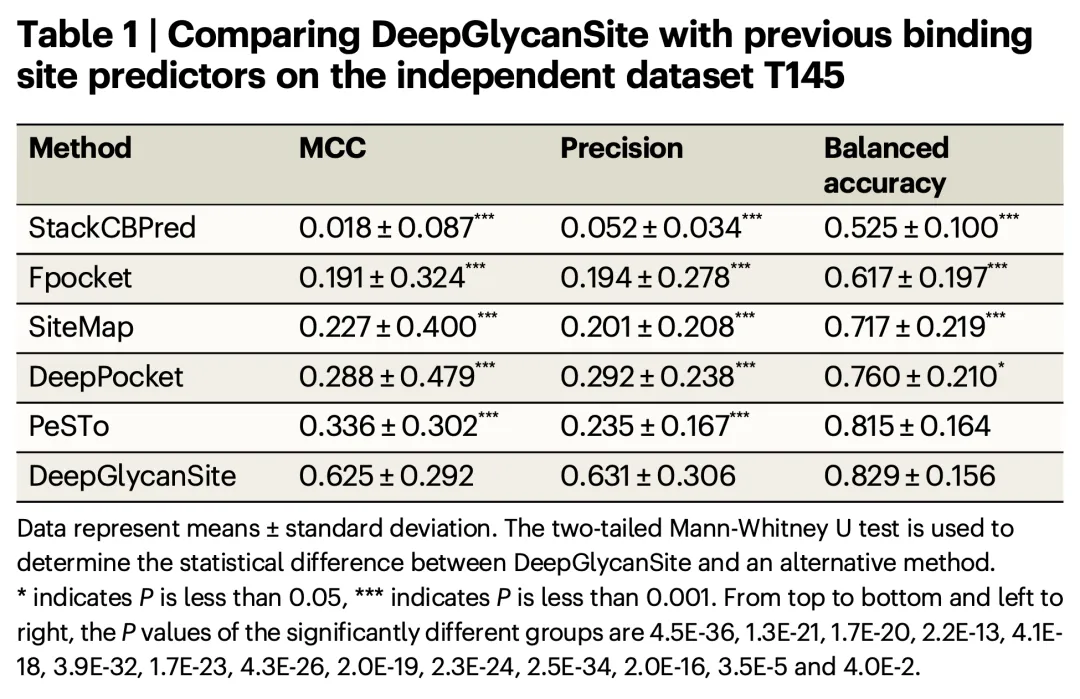
The study constructed a large dataset containing approximately 8,100 proteins and 1,700 carbohydrates and evaluated the performance of the DeepGlycanSite model on multiple independent test sets. The results demonstrated that DeepGlycanSite outperforms existing methods in detecting carbohydrate-binding sites.
Key Metrics:
Matthew’s Correlation Coefficient (MCC): 0.625 (average on independent test sets)
Precision: 0.631
Balanced Accuracy: 0.829
These metrics significantly exceed those of other comparison methods, highlighting the superior performance of DeepGlycanSite.
Conclusion
DeepGlycanSite is a highly efficient prediction tool that leverages Uni-Mol’s robust molecular representation capabilities to enhance the accuracy of carbohydrate-binding site predictions on proteins. By integrating sequence and structural information, DeepGlycanSite not only surpasses traditional methods in detecting monosaccharide or disaccharide binding sites but also excels in identifying multiple binding sites. This provides critical insights into carbohydrate-protein interactions.
Uni-Mol's ability to precisely capture chemical features and significantly improve predictive performance has established DeepGlycanSite as a powerful tool for addressing complex biological tasks. Its low dependence on protein structural accuracy enables analysis using predicted structures, supporting research into carbohydrate biological functions and drug development.
The study encourages researchers to explore Uni-Mol for various downstream applications in different domains. The team welcomes collaboration and discussion to unlock further possibilities!
Reference: [1] He, X., Zhao, L., Tian, Y. et al. Highly accurate carbohydrate-binding site prediction with DeepGlycanSite. Nat Commun 15, 5163 (2024). https://doi.org/10.1038/s41467-024-49516-2 [2] Zhou G, Gao Z, Ding Q, Zheng H, Xu H, Wei Z, et al. Uni-Mol: A Universal 3D Molecular Representation Learning Framework. ChemRxiv. 2023; doi:10.26434/chemrxiv-2022-jjm0j-v4